FDA UDI Mandate
Starting in 2014, and concluding in 2018, all medical devices must follow FDA UDI requirements.
There are two components to the FDA UDI requirements.
- Maintain device information in central database hosted by the FDA.
- Comply with UDI Device Marking guidelines for all devices.
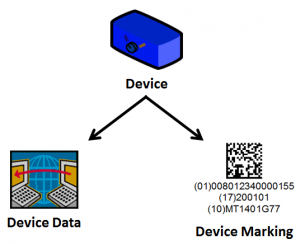
Both of these components work together to ensure accurate identification and traceability of medical devices.
Responsibility for meeting these requirements fall on what the FDA has defined as the “labeler”. Specifically, this is the “the person who causes a label to be applied to a device, or who causes the label to be modified, with the intent that the device will be introduced into interstate commerce without any subsequent replacement or modification of the label; in most instances, the labeler would be the device manufacturer, but the labeler may be a specification developer, a single-use device reprocessor, a convenience kit assembler, a repackager, or a relabeler.”
To comply, the FDA has recognized several acceptable standards: GS1, HIBCC, and ICCBBA. While each standard is different, they all share the ability to convey required UDI data in standardized data formats and standardized barcode formats.

